MOLECULAR DYNAMICS AND D.F.T.
OVERVIEW:
We are mainly interested in bridging some gap in multiscale computation
which has become more and more investigated in the last
decades. Multiscale computation is the sequence of simulations
from nanoscale quantum mechanics till macroscopic simulation.
This sequence usually consists of electronic structure, molecular
dynamics (MD), mesoscopic simulation, quasi-continuum, till FEM/CAD
(Finite Element Method/Computer Aided Design). The central component
that a user usually wishes to perfection in MD packages
is the local forces which are derived from
the potential energy. That is because all other parts of MD packets
are almost optimal: time integration, atom repositioning, etc.
There are numerous empirical potentials including Lennard-Jones,
Finnis-Sinclair, ReaxFF(Reactive Force Field), REBO(Reactive Empirical
Bond Order) and BOP(Bond Order Potential) among others. Our principal motivations
are the following items:
- Multiscale computation: implementation of bridges to allow
nanoscopic, mesoscopic till continuum media.
- Use DFT (Density Functional Theory) computations to generate potential
energy used in molecular dynamics.
- Getting acquainted with several usual molecular dynamics potential.
- Finite Element Method interface.
- Nanocomposites using polymers and carbon nanotube as nanofibers.
- Generation of potentials for special structures including graphene.
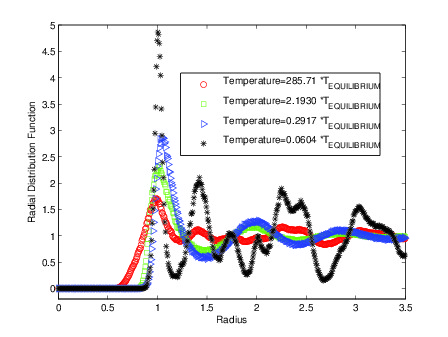
- Applications: effect of temperature, analysis of radial distributed
functions, etc.
Not only we are interested in usual potential energies in standard molecular
dynamics but we develop also method to obtain more accurate potential energies.
We use different computational tools to compute DFT, MD and FEM applications.
Those potentials should come from DFT and they will subsequently be
incorporated in MD packages.

GALLERY:
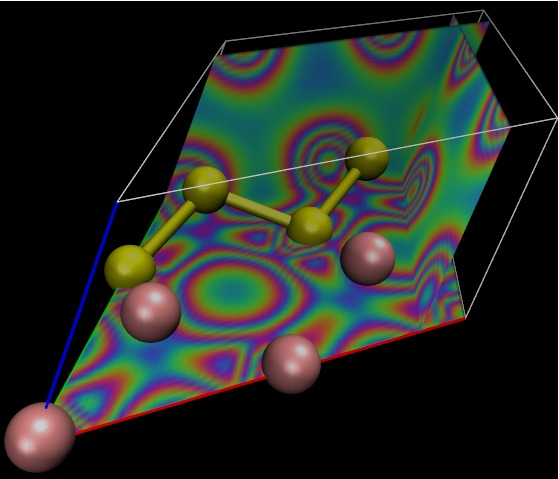
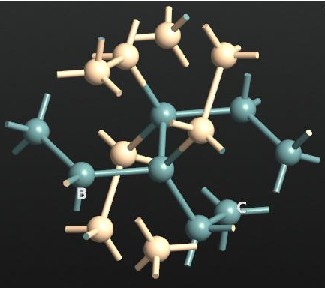
DFT (Germanium/Silicon)
|
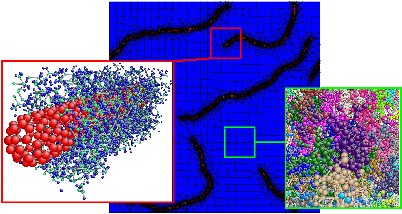
SELF-ASSEMBLY

|
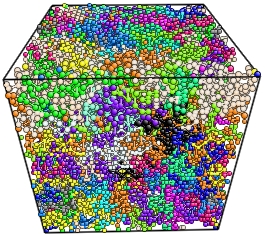
NANOCOMPOSITE
|
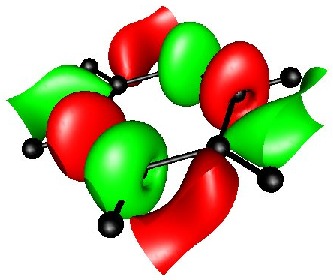
BENZENE ISOSURFACE
|
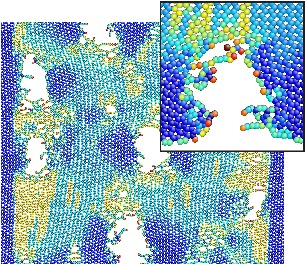
GRAPHENE FRACTURE
|
GexSi1-x
|
R.D.F.
|
POLYMER MATRIX
|


